























 放大字體
放大字體 縮小字體
縮小字體
57歲賴先生,常常運動,近年有手腳漸漸麻木、無力和針刺痛,沒辦法爬上樓梯,還有胃腸不適、便秘和拉肚子的問題困擾,他家族中的一個堂叔也有類似的症狀,經由抽血基因檢查後才發現是家族型類澱粉神經病變。
什麼是家族型類澱粉神經病變?
家族型類澱粉神經病變(Familial Amyloidotic Polyneuropathy,FAP)是一種罕見的體染色體顯性遺傳的疾病,不正常澱粉樣蛋白(amyloid)堆積在腦膜、心臟、眼睛、肺、腸胃道系統,影響周邊神經及自主神經系統等。在台灣推估有100~150個家族有此疾病,發病的年齡在男性約55歲,,女性約65歲。在台灣的案例,主要是侵犯周邊和心臟,早期的症狀為腕隧道症候群。
這個病變主要會影響轉甲狀腺素(transthyretin)基因突變,導致結構改變,類澱粉析維囤積在人體的器官組織上,因為無法有效清除澱粉沉積,所以有50%的機會被遺傳給下一代。
家族型類澱粉神經病變的診斷和治療

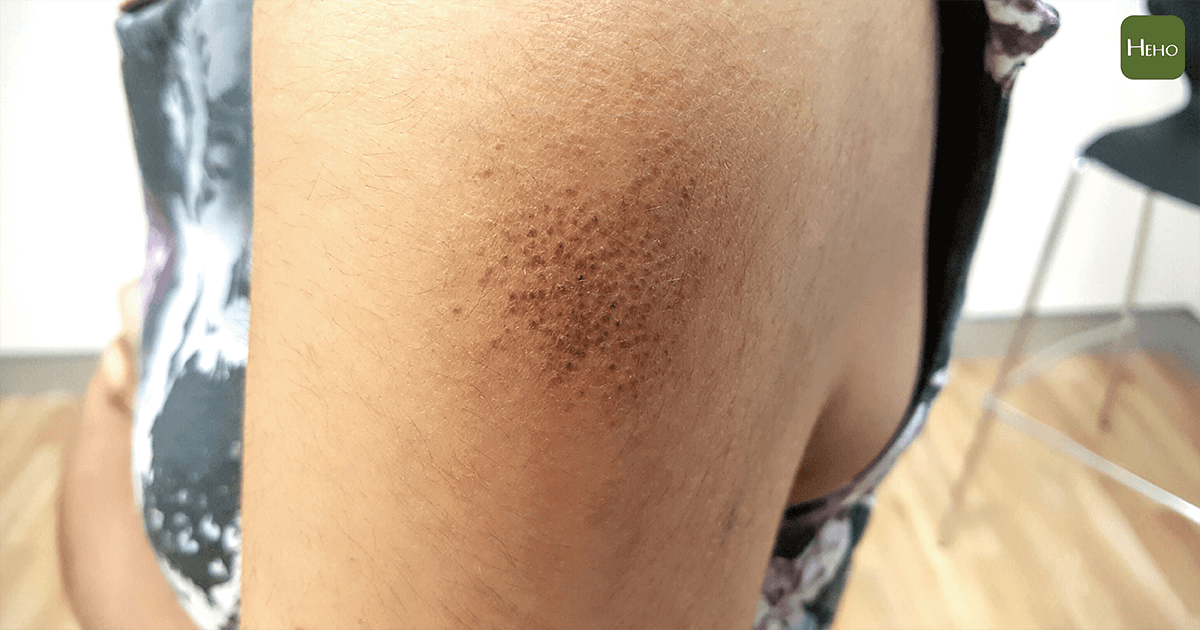
診斷此疾病,除了臨床表徵,家族病史,檢驗方面,如果侵犯周邊神經病變,可以安排神經傳導及肌電圖的檢查,評估它的嚴重度,心臟方面,可安排心臟超音波或核醫的檢查作評量,病理的檢查可作神經切片,直腸內膜切片,或肚皮下的脂肪切片,標本染剛果紅,如果有類澱粉囤積在組織上,在顯微鏡下會呈現蘋果綠偏光。
家族性類澱粉沉積病變的患者,會因為類澱粉沉積,導致組織和器官一直逐漸退化,甚至致死。過去的治療方式只能透過換肝,把會製造類澱粉的肝臟換成不會製造成類澱粉的肝臟,若接受肝臟移植手術預後良好,換肝10年存活率為80%,沒換肝為30%,但捐贈的肝臟取得不易,而且還會有人體排斥的問題,很多病患後來也只能以以藥物治療為主。
北榮周邊神經科李宜中主任表示,過去沒有有效的治療方法只能換肝,小分子干擾核糖核酸的新藥,可降低病人血中的突變轉甲狀腺素蛋白的產生及囤積,有效控制並延緩病情。北榮和國際發明小分子機轉藥物團隊,讓賴先生餐與
參與此臨床研究,每3週接受注射一次,原本突變的轉甲狀腺素蛋白降低至20%以下,目前恢復良好,研究成果發表在2018年7月的《The New England Journal of Medicine,NEJM》(新英格蘭醫學雜誌期刊)刊登,是治療此類遺傳疾病的一線希望。

北榮周邊神經科醫師林恭平表示,此罕見疾病的基因遺傳率為50%,但有這種體質不一定會生病,在各國的發病率不同,例如:葡萄牙圖變率為93%、瑞典和法國為60%,國內目前的發生率還需要更多要本來確認,但新的治療方法,可已讓病人在有早期疾病徵兆時,就可得到積極的治療。此外,關於如何預防遺傳疾病的問題,李宜中說,目前可以使用胚胎著床前的基因檢測,搭配人工生殖試管嬰兒的方法,來培育沒有疾病的孩子。
延伸閱讀
心臟只有拇指大,體重只有1948克早產兒患罕見心臟病,馬偕神救援
文/黃聖筑 圖/許嘉真